Sequence-structure-function characterization of the emerging tetracycline destructases family of antibiotic resistance enzymes
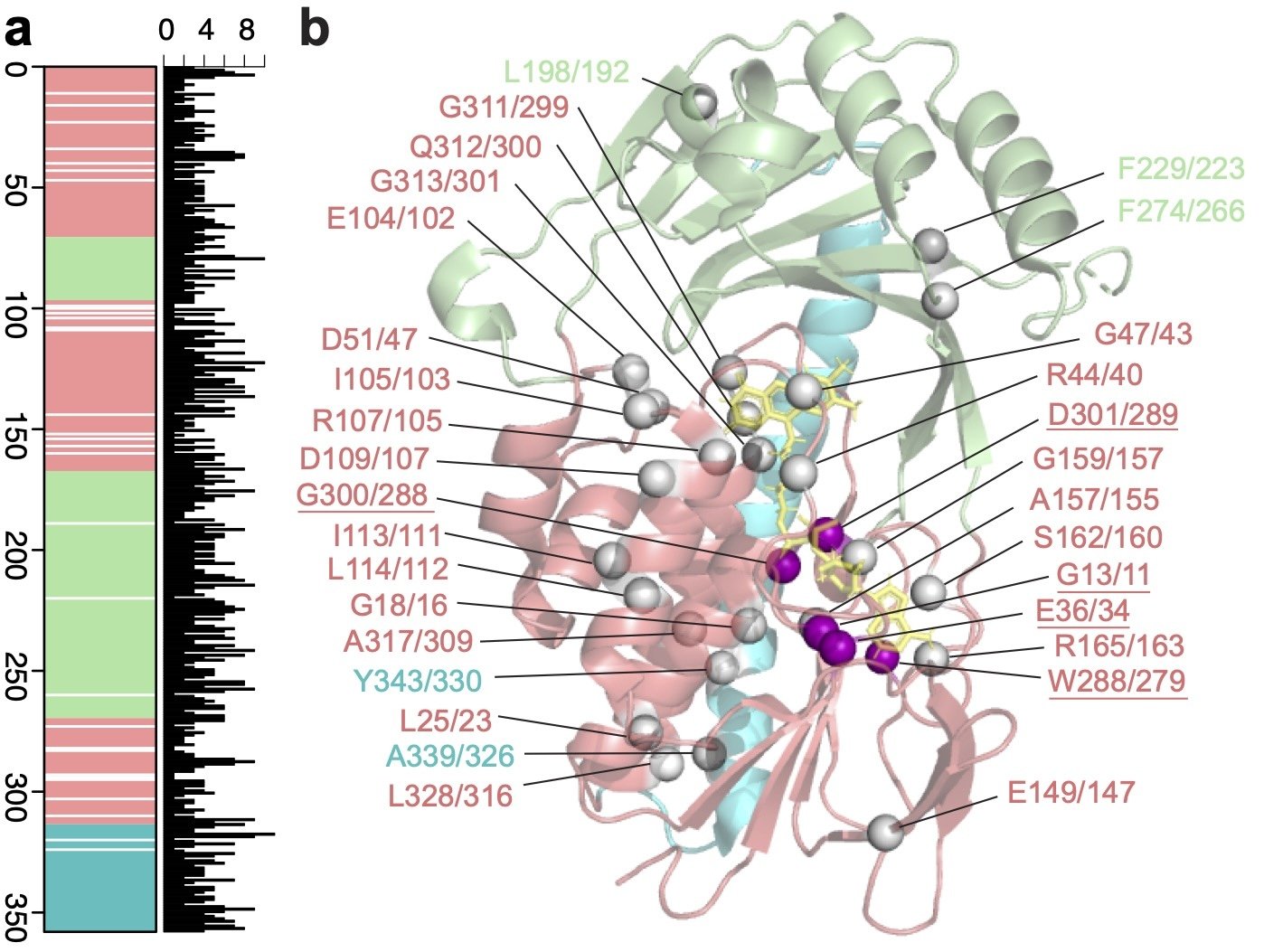
Figure 1. Identification of amino acid positions essential to TDase function. a Linear geneplot of TDase MSA. White lines indicate a 100% conserved position. b The Tet(X7) structure (PDB 6WG9) with the 31 conserved residues shown as spheres. Spheres for true essential residues are colored purple, while conditionally essential residues are white.
Like human-made machines, the functions of protein machines are determined by their structure. This is why hypodermic syringes and bacterial type III secretion systems, both of which inject materials past barriers, have remarkably similar structures. A fundamental difference, however, is that while the shape of human-made machines comes from the bolting and fusing of plastic and metal parts, proteins are shaped by the intricate twisting and folding of linear amino acids chains. Thus, a protein’s amino acid sequence determines its structure, which in turn determines its function—a principle dubbed the “sequence-structure-function” paradigm.
We leveraged this to better understand the sequence determinants of function for the tetracycline destructase (TDase) family of antibiotic resistance enzymes. Tetracycline antibiotics are over 70 years old, and are widely used in agriculture and the clinic. TDases are flavoenzymes that covalently modify and inactivate tetracycline molecules, eliminating their antibiotic activity and allowing bacteria to survive antibiotic treatment 1,2. For decades, the TDases were relatively unknown and uncommon, but that changed between 2015 and 2020 when a flurry of reports described the discovery of >30 new TDase sequences from pathogens around the world. In the blink of an eye, TDases went from interesting oddities to urgent clinical threats.
This wealth of new TDase sequence data permitted us to engage in a deep investigation of the TDase sequence-structure-function landscape. We approached this using two complementary methods:
Discovering new TDases with profile HMMs
First, we constructed profile hidden Markov models (HMM) to retrospectively search for TDases in public sequence databases. We reasoned many uncharacterized TDases could be hiding in older genomic and metagenomic sequencing projects, having been sequenced but not identified as TDases because nobody looked for them. Profile HMMs are probabilistic models that capture the conservation of each amino acid at each position of a multiple sequence alignment. This makes them ideally suited to identify remote homologs with low overall sequence identity.
Using these, we identified 50 high-scoring sequences likely to be TDases. We inserted these into an Escherichia coli host and screened these strains with tetracycline. Since the E. coli is natively sensitive to tetracycline, any strain which could could grow on the tetracycline must be because of the inserted HMM-predicted sequence, validating it as a functional TDases. In total we discovered 13 new TDases, with sequence identities as low as 55% to other known enzymes. Many belonged to a sub-clade of TDases that we traced back to Legionella species genomes, including the pathogenic L. longbeachae. We solved the X-ray crystal structures of two Legionella TDases, Tet(56-2) and Tet(56-3), and compared these to the already solved structure of the related Tet(56) 3. This revealed several important insights into this clade, including that the “gate-keeper” helix of the Tet(56)-like structures orients similarly to eachother, but differently from other TDases. Further, a loop which was disordered in the Tet(56) structure had a distinct confirmation in Tet(56-2),oritented away from the substrate-binding channel, which suggests an important role in active site gating.
Identifying amino acid residues essential to function
The sequence-structure-paradigm predicts that amino acids which are critical for enzyme function will be conserved across protein families, as any substitution will decrease function. Therefore, for our second approach, we used the sequence conservation captured by our HMMs to identify amino acid positions important to TDase activity . By analyzing 114 functional TDase variants, we identified 31 positions 100% conserved across the entire protein family (Figure 1).
To phenotypically evaluate the essentiality of each conserved position, we performed alanine-scanning mutagenesis on two parent TDases, Tet(X7) and Tet(50), at 27 conserved positions. (The remaining positions were alanine already.) If a mutant lost all function against a tetracycline antibiotic then the position is essential. We observed that mutations at all positions resulted in some decrease in activity. 5/27 were “true essential,” with mutations in both proteins resulting in complete loss of activity to all tetracyclines. The remaining 22 positions were “conditionally essential,” meaning they completely lost activity to some drugs in at least one of the protein backgrounds, but still maintained some activity in others—albeit at degrees lower than the parent TDase. Notably, this structure-independent approach re-identified residues previously shown to be essential using structural analyses, such as the GxGxxG flavin-binding motif, while also identifying new ones.
Conclusion
In this report, we used complementary sequence-based approaches to conduct a broad survey of protein sequences for TDase activity, and engage in a deep-dive into the sequence-structure function of TDase enzymology. Our TDase HMMs enabled more accurate scoring and prediction of functional TDases which could be used for future antibiotic resistance surveillance efforts. And our identification of key residues important for TDase function provide valuable insights into the workings of these enzymes which could inform strategies that interfere with that function and rescue tetracycline antibiotics. Taken together, we have set the foundation for future work focusing on the specific interactions and functional roles of each residue and how they influence TDases’ tetracycline-inactivating activity.
References
Forsberg KJ, Patel S, Wencewicz TA, Dantas G. The Tetracycline Destructases: A Novel Family of Tetracycline-Inactivating Enzymes. Chem Biol. 2015 Jul 23;22(7):888-97. doi: 10.1016/j.chembiol.2015.05.017. Epub 2015 Jun 18. PMID: 26097034; PMCID: PMC4515146.
Gasparrini AJ, Markley JL, Kumar H, Wang B, Fang L, Irum S, Symister CT, Wallace M, Burnham CD, Andleeb S, Tolia NH, Wencewicz TA, Dantas G. Tetracycline-inactivating enzymes from environmental, human commensal, and pathogenic bacteria cause broad-spectrum tetracycline resistance. Commun Biol. 2020 May 15;3(1):241. doi: 10.1038/s42003-020-0966-5. PMID: 32415166; PMCID: PMC7229144.
Park J, Gasparrini AJ, Reck MR, Symister CT, Elliott JL, Vogel JP, Wencewicz TA, Dantas G, Tolia NH. Plasticity, dynamics, and inhibition of emerging tetracycline resistance enzymes. Nat Chem Biol. 2017 Jul;13(7):730-736. doi: 10.1038/nchembio.2376. Epub 2017 May 8. PMID: 28481346; PMCID: PMC5478473.